
Novel Anti-Melanogenic Compounds, (Z)-5-(Substituted Benzylidene)-4-thioxothiazolidin-2-one Derivatives: In Vitro and In Silico Insights

 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Results and Discussion
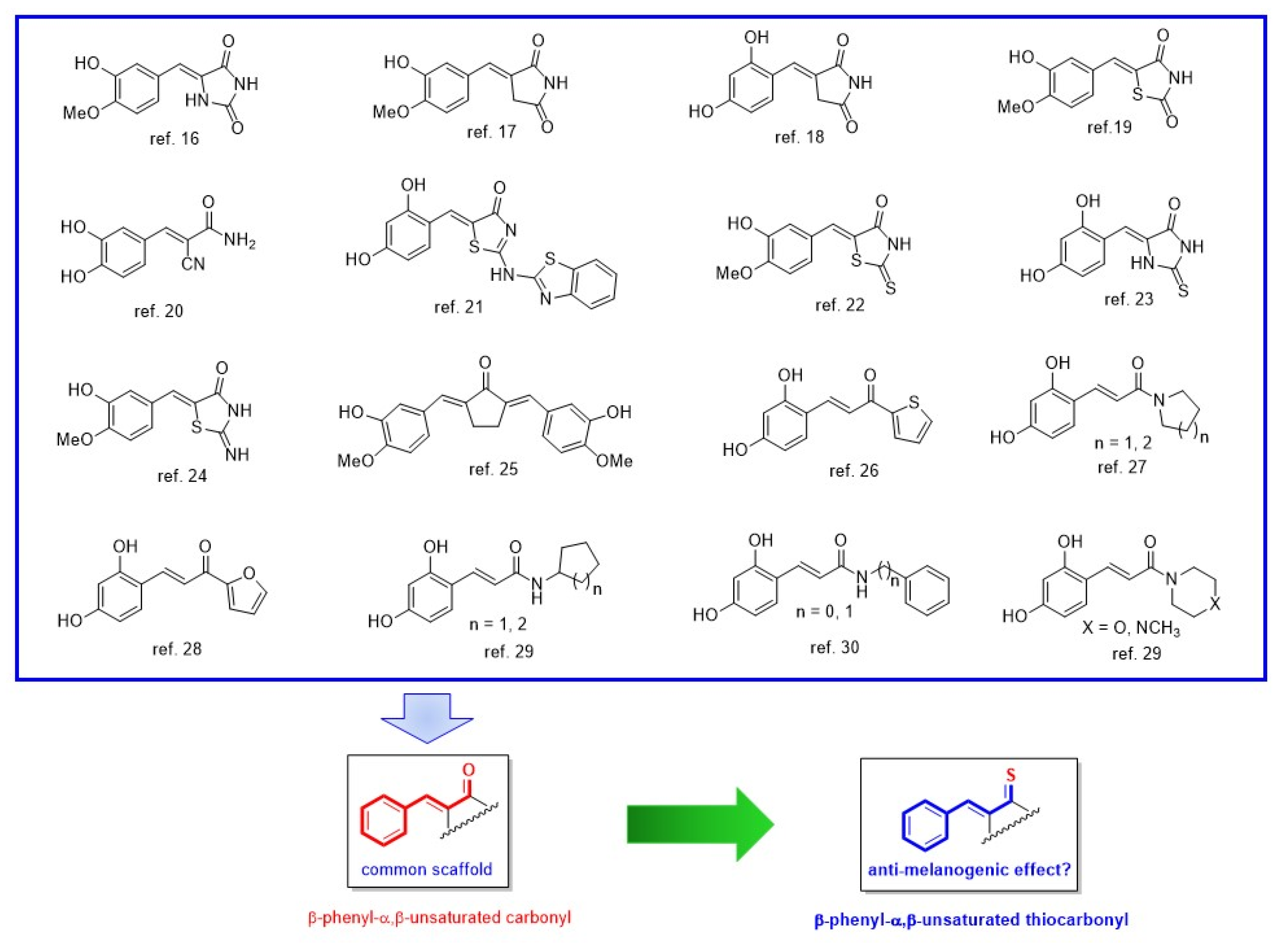
2.1. Chemistry
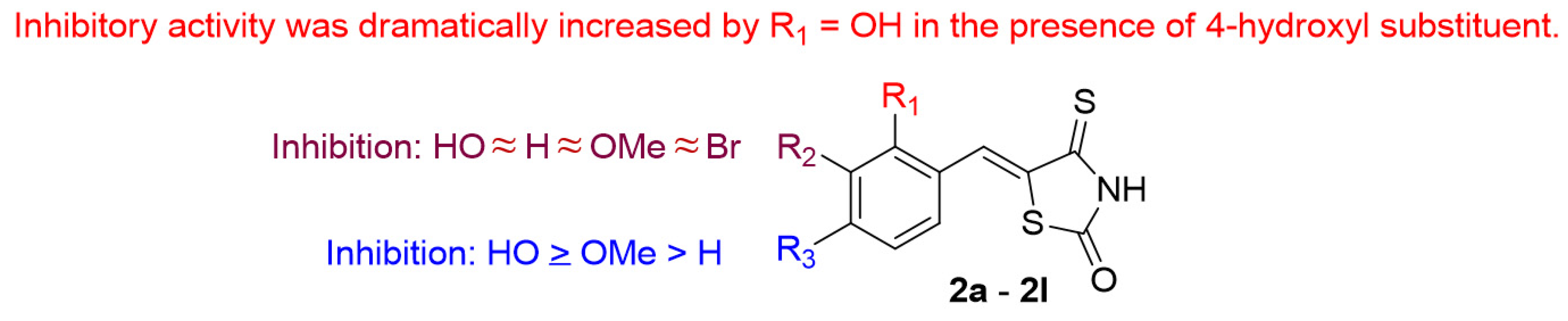
2.2. Mushroom Tyrosinase Inhibition and Values of Logarithm of Partition Function
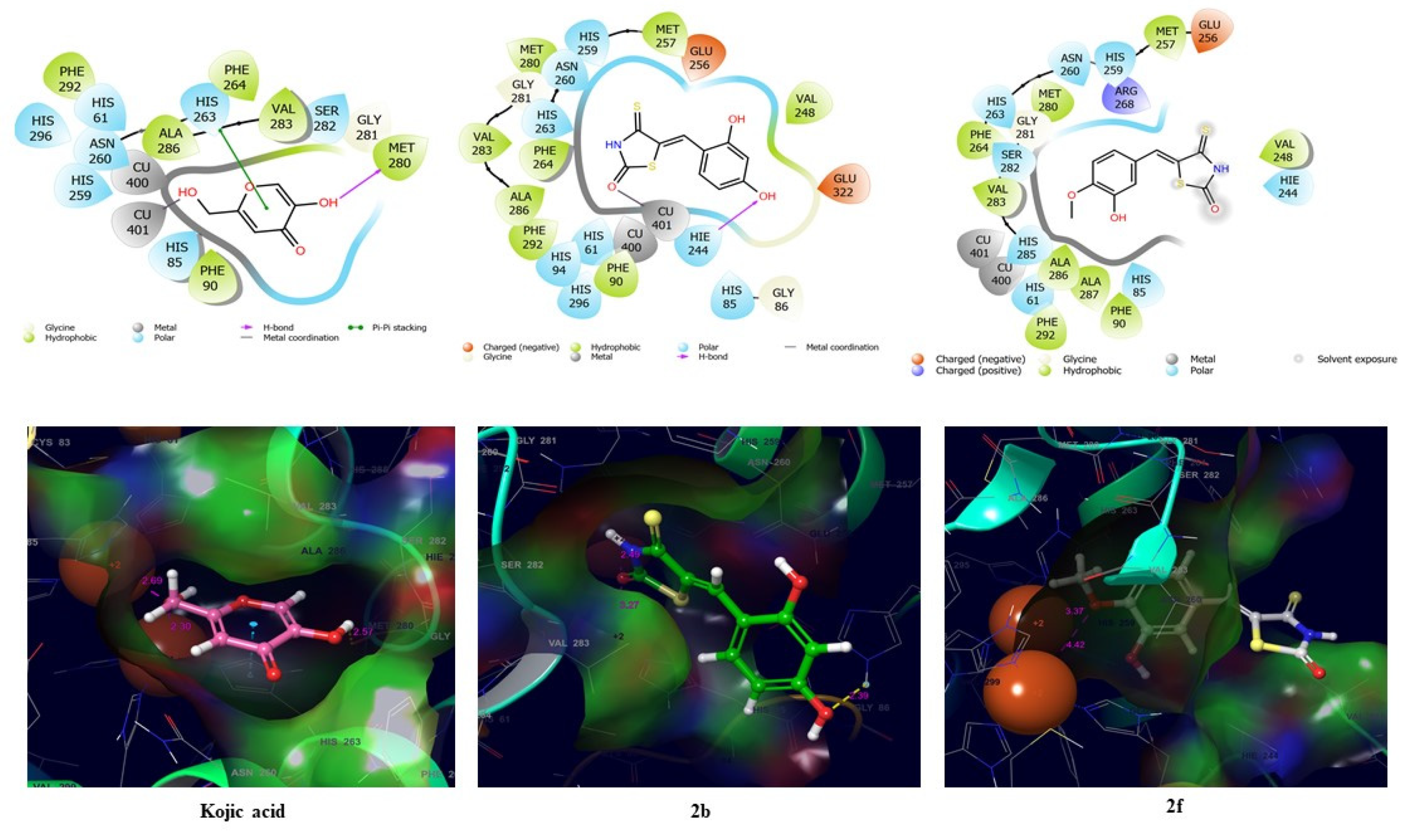
2.3. Modes of Action of Compounds 2b and 2f
2.4. In Silico Studies of (Z)-BTTZ Derivatives 2b and 2f
2.4.1. Docking Simulations of Compounds 2b and 2f and Kojic Acid with Mushroom Tyrosinase
2.4.2. Docking Simulation of Compounds 2b and 2f and Kojic Acid with the Human Tyrosinase Homology Model
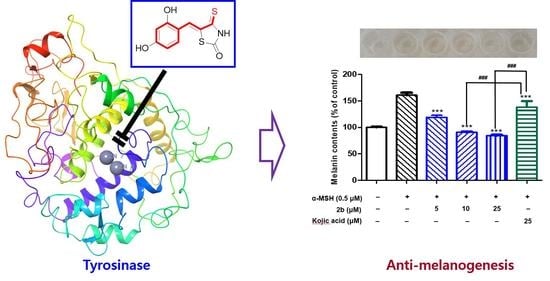
2.5. Cytotoxic Effects of Compounds 2b and 2f
2.6. Anti-Melanogenic Activities of Compounds 2b and 2f
2.7. Anti-Tyrosinase Activities of Compounds 2b and 2f
3. Conclusions
4. Materials and Methods
4.1. Chemistry
4.1.1. General Methods
Synthesis of 4-thioxothiazolidin-2-one (1)
General Procedure Used to Synthesize (Z)-5-(Substituted Benzylidene)-4-thioxothiazolidin-2-one Derivatives (Compounds 2a–2l)
(Z)-5-(4-Hydroxybenzylidene)-4-thioxothiazolidin-2-one (Compound 2a)
(Z)-5-(2,4-Dihydroxybenzylidene)-4-thioxothiazolidin-2-one (Compound 2b)
(Z)-5-(3,4-Dihydroxybenzylidene)-4-thioxothiazolidin-2-one (Compound 2c)
(Z)-5-(4-Hydroxy-3-methoxybenzylidene)-4-thioxothiazolidin-2-one (compound 2d)
(Z)-5-(3-Ethoxy-4-hydroxybenzylidene)-4-thioxothiazolidin-2-one (Compound 2e)
(Z)-5-(3-Hydroxy-4-methoxybenzylidene)-4-thioxothiazolidin-2-one (Compound 2f)
(Z)-5-(4-Hydroxy-3,5-dimethoxybenzylidene)-4-thioxothiazolidin-2-one (Compound 2g)
(Z)-5-(2-Hydroxybenzylidene)-4-thioxothiazolidin-2-one (Compound 2h)
(Z)-5-(3-Bromo-4-hydroxybenzylidene)-4-thioxothiazolidin-2-one (Compound 2i)
(Z)-5-(4-Methoxybenzylidene)-4-thioxothiazolidin-2-one (Compound 2j)
(Z)-5-(2,4-Dimethoxybenzylidene)-4-thioxothiazolidin-2-one (Compound 2k)
(Z)-5-(3,4-Dimethoxybenzylidene)-4-thioxothiazolidin-2-one (Compound 2l)
4.2. Tyrosinase Inhibition—Kinetic and In Silico Studies
4.2.1. Mushroom Tyrosinase Inhibition Assay
4.2.2. Kinetic Studies on Mushroom Tyrosinase Inhibition by 2b and 2f
4.2.3. In Silico Study of Interactions between Mushroom Tyrosinase and Compounds 2b and 2f and Kojic Acid
4.2.4. In Silico Study of Interactions between Compounds 2b and 2f and Kojic Acid and the Human Tyrosinase Homology Model
4.3. Cell Culture
4.4. Cell Viability Assays
4.5. Anti-Melanogenesis Activity Assays
4.6. Anti-Tyrosinase Activity Assays
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Naidoo, L.; Khoza, N.; Dlova, N.C. A Fairer Face, a Fairer Tomorrow? A Review of Skin Lighteners. Cosmetics 2016, 3, 33. [Google Scholar] [CrossRef]
- Sagoe, D.; Pallesen, S.; Dlova, N.C.; Lartey, M.; Ezzedine, K.; Dadzie, O. The global prevalence and correlates of skin bleaching: A meta-analysis and meta-regression analysis. Int. J. Dermatol. 2019, 58, 24–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Global Industry Analysts (GIA) Report. Available online: https://cosmetics.specialchem.com/news/industry-news/skin-lightening-products-market-report-000219549 (accessed on 4 April 2021).
- Maymone, M.B.C.; Neamah, H.H.; Wirya, S.A.; Patzelt, N.M.; Secemsky, E.A.; Zancanaro, P.Q.; Vashi, N.A. The impact of skin hyperpigmentation and hyperchromia on quality of life: A cross-sectional study. J. Am. Acad. Dermatol. 2017, 77, 775–778. [Google Scholar] [CrossRef]
- Ogiwara, Y.; Sugiura, M.; Watanabe, K.; Tawara, J.; Endo, E.; Maruyama, H.; Tsuji, S.; Matsue, K.; Yamada, H.; Wako, Y. Evaluation of the repeated-dose liver, bone marrow and peripheral blood micronucleus and comet assays using kojic acid. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 780, 111–116. [Google Scholar] [CrossRef]
- Westerhof, W.; Kooyers, T. Hydroquinone and its analogues in dermatology—A potential health risk. J. Cosmet. Dermatol. 2005, 4, 55–59. [Google Scholar] [CrossRef] [Green Version]
- Gaskell, M.; McLuckie, K.I.; Farmer, P.B. Genotoxicity of the benzene metabolites para-benzoquinone and hydroquinone. Chem.-Biol. Interact. 2005, 153, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Fukuda, M. Arbutin: Mechanism of its depigmenting action in human melanocyte culture. J. Pharmacol. Exp. Ther. 1996, 276, 765–769. [Google Scholar]
- Andersen, F.A.; Bergfeld, W.F.; Belsito, D.V.; Hill, R.A.; Klaassen, C.D.; Liebler, D.C.; Marks, J.G.; Shank, R.C.; Slaga, T.J.; Snyder, P.W. Final Amended Safety Assessment of Hydroquinone as Used in Cosmetics. Int. J. Toxicol. 2010, 29, 274S–287S. [Google Scholar] [CrossRef] [PubMed]
- Nordlund, J.; Grimes, P.; Ortonne, J. The safety of hydroquinone. J. Eur. Acad. Dermatol. Venereol. 2006, 20, 781–787. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Namasivayam, V.; Manickam, M.; Jung, S.-H. Inhibitors of Melanogenesis: An Updated Review. J. Med. Chem. 2018, 61, 7395–7418. [Google Scholar] [CrossRef]
- Chang, T.-S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef] [Green Version]
- Zolghadri, S.; Bahrami, A.; Hassan Khan, M.T.; Munoz-Munoz, J.; Garcia-Molina, F.; Garcia-Canovas, F.; Saboury, A.A. A comprehensive review on tyrosinase inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 279–309. [Google Scholar] [CrossRef] [Green Version]
- Ullah, S.; Son, S.; Yun, H.Y.; Kim, D.H.; Chun, P.; Moon, H.R. Tyrosinase inhibitors: A patent review (2011–2015). Expert Opin. Ther. Pat. 2016, 26, 347–362. [Google Scholar] [CrossRef]
- Roulier, B.; Pérès, B.; Haudecoeur, R. Advances in the Design of Genuine Human Tyrosinase Inhibitors for Targeting Melanogenesis and Related Pigmentations. J. Med. Chem. 2020, 63, 13428–13443. [Google Scholar] [CrossRef]
- Ha, Y.M.; Kim, J.-A.; Park, Y.J.; Park, D.; Kim, J.M.; Chung, K.W.; Lee, E.K.; Park, J.Y.; Lee, J.Y.; Lee, H.J. Analogs of 5-(substituted benzylidene) hydantoin as inhibitors of tyrosinase and melanin formation. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2011, 1810, 612–619. [Google Scholar] [CrossRef]
- Ha, Y.M.; Kim, J.-A.; Park, Y.J.; Park, D.; Choi, Y.J.; Kim, J.M.; Chung, K.W.; Han, Y.K.; Park, J.Y.; Lee, J.Y. Synthesis and biological activity of hydroxybenzylidenyl pyrrolidine-2, 5-dione derivatives as new potent inhibitors of tyrosinase. MedChemComm 2011, 2, 542–549. [Google Scholar] [CrossRef]
- Chung, K.W.; Park, Y.J.; Choi, Y.J.; Park, M.H.; Ha, Y.M.; Uehara, Y.; Yoon, J.H.; Chun, P.; Moon, H.R.; Chung, H.Y. Evaluation of in vitro and in vivo anti-melanogenic activity of a newly synthesized strong tyrosinase inhibitor (E)-3-(2, 4 dihydroxybenzylidene) pyrrolidine-2, 5-dione (3-DBP). Biochim. Biophys. Acta (BBA)-Gen. Subj. 2012, 1820, 962–969. [Google Scholar] [CrossRef]
- Ha, Y.M.; Park, Y.J.; Kim, J.-A.; Park, D.; Park, J.Y.; Lee, H.J.; Lee, J.Y.; Moon, H.R.; Chung, H.Y. Design and synthesis of 5-(substituted benzylidene) thiazolidine-2, 4-dione derivatives as novel tyrosinase inhibitors. Eur. J. Med. Chem. 2012, 49, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Son, S.; Kim, H.; Yun, H.Y.; Ullah, S.; Kim, S.J.; Kim, Y.-J.; Kim, M.-S.; Yoo, J.-W.; Chun, P.; Moon, H.R. (E)-2-Cyano-3-(substituted phenyl) acrylamide analogs as potent inhibitors of tyrosinase: A linear β-phenyl-α, β-unsaturated carbonyl scaffold. Bioorg. Med. Chem. 2015, 23, 7728–7734. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.H.; Lee, B.; Son, S.; Yun, H.Y.; Moon, K.M.; Jeong, H.O.; Kim, D.H.; Lee, E.K.; Choi, Y.J.; Do, H.K. (Z)-2-(Benzo [d] thiazol-2-ylamino)-5-(substituted benzylidene) thiazol-4 (5H)-one Derivatives as Novel Tyrosinase Inhibitors. Biol. Pharm. Bull. 2015, 38, 1227–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, E.; Lee, E.K.; Noh, S.G.; Jung, H.J.; Moon, K.M.; Park, M.H.; Park, Y.J.; Hyun, M.K.; Lee, A.K.; Kim, S.J. In vitro and in vivo evidence of tyrosinase inhibitory activity of a synthesized (Z)-5-(3-hydroxy-4-methoxybenzylidene)-2-thioxothiazolidin-4-one (5-HMT). Exp. Dermatol. 2019, 28, 734–737. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.R.; Lee, H.J.; Choi, Y.J.; Park, Y.J.; Woo, Y.; Kim, S.J.; Park, M.H.; Lee, H.W.; Chun, P.; Chung, H.Y. Benzylidene-linked thiohydantoin derivatives as inhibitors of tyrosinase and melanogenesis: Importance of the β-phenyl-α, β-unsaturated carbonyl functionality. MedChemComm 2014, 5, 1410–1417. [Google Scholar] [CrossRef]
- Jung, H.J.; Lee, M.J.; Park, Y.J.; Noh, S.G.; Lee, A.K.; Moon, K.M.; Lee, E.K.; Bang, E.J.; Park, Y.J.; Kim, S.J. A novel synthetic compound,(Z)-5-(3-hydroxy-4-methoxybenzylidene)-2-iminothiazolidin-4-one (MHY773) inhibits mushroom tyrosinase. Biosci. Biotechnol. Biochem. 2018, 82, 759–767. [Google Scholar] [CrossRef]
- Jung, H.J.; Lee, A.K.; Park, Y.J.; Lee, S.; Kang, D.; Jung, Y.S.; Chung, H.Y.; Moon, H.R. (2E, 5E)-2, 5-Bis (3-hydroxy-4-methoxybenzylidene) cyclopentanone exerts anti-melanogenesis and anti-wrinkle activities in B16F10 melanoma and Hs27 fibroblast cells. Molecules 2018, 23, 1415. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.S.; Noh, S.G.; Park, Y.; Kang, D.; Chun, P.; Chung, H.Y.; Jung, H.J.; Moon, H.R. A potent tyrosinase inhibitor,(E)-3-(2, 4-Dihydroxyphenyl)-1-(thiophen-2-yl) prop-2-en-1-one, with anti-melanogenesis properties in α-MSH and IBMX-induced B16F10 melanoma cells. Molecules 2018, 23, 2725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, S.; Park, Y.; Ikram, M.; Lee, S.; Park, C.; Kang, D.; Yang, J.; Akter, J.; Yoon, S.; Chun, P. Design, synthesis and anti-melanogenic effect of cinnamamide derivatives. Bioorg. Med. Chem. 2018, 26, 5672–5681. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Noh, S.G.; Ryu, I.Y.; Park, C.; Lee, J.Y.; Chun, P.; Moon, H.R.; Chung, H.Y. (E)-1-(Furan-2-yl)-(substituted phenyl) prop-2-en-1-one Derivatives as Tyrosinase Inhibitors and Melanogenesis Inhibition: An In Vitro and In Silico Study. Molecules 2020, 25, 5460. [Google Scholar] [CrossRef]
- Ullah, S.; Park, C.; Ikram, M.; Kang, D.; Lee, S.; Yang, J.; Park, Y.; Yoon, S.; Chun, P.; Moon, H.R. Tyrosinase inhibition and anti-melanin generation effect of cinnamamide analogues. Bioorg. Chem. 2019, 87, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Ullah, S.; Kang, D.; Lee, S.; Ikram, M.; Park, C.; Park, Y.; Yoon, S.; Chun, P.; Moon, H.R. Synthesis of cinnamic amide derivatives and their anti-melanogenic effect in α-MSH-stimulated B16F10 melanoma cells. Eur. J. Med. Chem. 2019, 161, 78–92. [Google Scholar] [CrossRef]
- Tripathi, A.C.; Gupta, S.J.; Fatima, G.N.; Sonar, P.K.; Verma, A.; Saraf, S.K. 4-Thiazolidinones: The advances continue…. Eur. J. Med. Chem. 2014, 72, 52–77. [Google Scholar] [CrossRef]
- Lesyk, R.B.; Zimenkovsky, B.S. 4-Thiazolidones: Centenarian History, Current Status and Perspectives for Modern Organic and Medicinal Chemistry. Curr. Org. Chem. 2004, 8, 1547–1577. [Google Scholar] [CrossRef]
- Tomašić, T.; Peterlin Mašič, L. Rhodanine as a scaffold in drug discovery: A critical review of its biological activities and mechanisms of target modulation. Expert Opin. Drug Discov. 2012, 7, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Maddila, S.; Gorle, S.; Jonnalagadda, S.B. Drug screening of rhodanine derivatives for antibacterial activity. Expert Opin. Drug Discov. 2020, 15, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Vaidya, A.; Ravichandran, V.; Kashaw, S.K.; Agrawal, R.K. Recent developments and biological activities of thiazolidinone derivatives: A review. Bioorg. Med. Chem. 2012, 20, 3378–3395. [Google Scholar] [CrossRef] [PubMed]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. Thiazolidine-2,4-diones as multi-targeted scaffold in medicinal chemistry: Potential anticancer agents. Eur. J. Med. Chem. 2014, 87, 814–833. [Google Scholar] [CrossRef]
- Lesyk, R.; Zimenkovsky, B.; Atamanyuk, D.; Jensen, F.; Kieć-Kononowicz, K.; Gzella, A. Anticancer thiopyrano[2,3-d][1,3]thiazol-2-ones with norbornane moiety. Synthesis, cytotoxicity, physico-chemical properties, and computational studies. Bioorg. Med. Chem. 2006, 14, 5230–5240. [Google Scholar] [CrossRef]
- Vogeli, U.; Philipsborn, W.V.; Nagarajan, K.; Nair, M.D. C-13-NMR Spectroscopy Part 19. Structures of Addition-Products of Acetylene-Dicarboxylic Acid-Esters with Various Dinucleophiles—Application of C,H-Spin-Coupling Constants. Helv. Chim. Acta 1978, 61, 607–617. [Google Scholar] [CrossRef]
- Slominski, A.; Zmijewski, M.A.; Pawelek, J. L-Tyrosine and L-dihydroxyphenylalanine as hormone-like regulators of melanocyte functions. Pigment Cell Melanoma Res. 2012, 25, 14–27. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef]
- Lai, X.; Wichers, H.J.; Soler-Lopez, M.; Dijkstra, B.W. Structure of Human Tyrosinase Related Protein 1 Reveals a Binuclear Zinc Active Site Important for Melanogenesis. Angew. Chem. Int. Ed. 2017, 56, 9812–9815. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [Green Version]
- Hyun, S.K.; Lee, W.-H.; Jeong, D.M.; Kim, Y.; Choi, J.S. Inhibitory effects of kurarinol, kuraridinol, and trifolirhizin from Sophora flavescens on tyrosinase and melanin synthesis. Biol. Pharm. Bull. 2008, 31, 154–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, I.Y.; Choi, I.; Jung, H.J.; Ullah, S.; Choi, H.; Al-Amin, M.; Chun, P.; Moon, H.R. In vitro anti-melanogenic effects of chimeric compounds, 2-(substituted benzylidene)-1,3-indanedione derivatives with a β-phenyl-α, β -unsaturated dicarbonyl scaffold. Bioorg. Chem. 2021, 109, 104688. [Google Scholar] [CrossRef]
- Hassan, M.; Ashraf, Z.; Abbas, Q.; Raza, H.; Seo, S.-Y. Exploration of novel human tyrosinase inhibitors by molecular modeling, docking and simulation studies. Interdiscip. Sci. Comput. Life Sci. 2018, 10, 68–80. [Google Scholar] [CrossRef]
- Saeed, A.; Mahesar, P.A.; Channar, P.A.; Abbas, Q.; Larik, F.A.; Hassan, M.; Raza, H.; Seo, S.-Y. Synthesis, molecular docking studies of coumarinyl-pyrazolinyl substituted thiazoles as non-competitive inhibitors of mushroom tyrosinase. Bioorg. Chem. 2017, 74, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Larik, F.A.; Saeed, A.; Channar, P.A.; Muqadar, U.; Abbas, Q.; Hassan, M.; Seo, S.-Y.; Bolte, M. Design, synthesis, kinetic mechanism and molecular docking studies of novel 1-pentanoyl-3-arylthioureas as inhibitors of mushroom tyrosinase and free radical scavengers. Eur. J. Med. Chem. 2017, 141, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef]
- Kim, H.; Roh, H.S.; Kim, J.E.; Park, S.D.; Park, W.H.; Moon, J.-Y. Compound K attenuates stromal cell-derived growth factor 1 (SDF-1)-induced migration of C6 glioma cells. Nutr. Res. Pract. 2016, 10, 259. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.G.; Chang, W.L.; Lee, C.J.; Lee, L.T.; Shih, C.M.; Wang, C.C. Melanogenesis inhibition by gallotannins from Chinese galls in B16 mouse melanoma cells. Biol. Pharm. Bull. 2009, 32, 1447–1452. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.J.; Ha, Y.M.; Kim, J.A.; Park, J.Y.; Ha, T.K.; Park, D.; Chun, P.; Park, N.H.; Moon, H.R.; Chung, H.Y. A novel synthesized tyrosinase inhibitor: (E)-2-((2,4-dihydroxyphenyl)diazenyl)phenyl 4-methylbenzenesulfonate as an azo-resveratrol analog. Biosci. Biotechnol. Biochem. 2013, 77, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | IC50 (µM) | a Log p |
| 2a | H | H | OH | H | 28.05 ± 1.16 | 1.65 |
| 2b | OH | H | OH | H | 0.47 ± 0.97 | 1.26 |
| 2c | H | OH | OH | H | 47.41 ± 2.18 | 1.26 |
| 2d | H | OMe | OH | H | 70.75 ± 1.00 | 1.52 |
| 2e | H | OEt | OH | H | 92.81 ± 0.89 | 1.86 |
| 2f | H | OH | OMe | H | 15.24 ± 1.68 | 1.52 |
| 2g | H | OMe | OH | OMe | 82.39 ± 1.26 | 1.39 |
| 2h | OH | H | H | H | 147.61 ± 0.96 | 1.65 |
| 2i | H | Br | OH | H | 26.27 ± 4.10 | 2.47 |
| 2j | H | H | OMe | H | 30.83 ± 1.41 | 1.91 |
| 2k | OMe | H | OMe | H | 126.35 ± 0.46 | 1.78 |
| 2l | H | OMe | OMe | H | 23.31 ± 0.28 | 1.78 |
| Kojic acid | 66.30 ± 0.75 | −2.45 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.; Ryu, I.Y.; Choi, I.; Ullah, S.; Jung, H.J.; Park, Y.; Jeong, Y.; Hwang, Y.; Hong, S.; Yoon, I.-S.; et al. Novel Anti-Melanogenic Compounds, (Z)-5-(Substituted Benzylidene)-4-thioxothiazolidin-2-one Derivatives: In Vitro and In Silico Insights. Molecules 2021, 26, 4963. https://doi.org/10.3390/molecules26164963
Choi H, Ryu IY, Choi I, Ullah S, Jung HJ, Park Y, Jeong Y, Hwang Y, Hong S, Yoon I-S, et al. Novel Anti-Melanogenic Compounds, (Z)-5-(Substituted Benzylidene)-4-thioxothiazolidin-2-one Derivatives: In Vitro and In Silico Insights. Molecules. 2021; 26(16):4963. https://doi.org/10.3390/molecules26164963
Chicago/Turabian StyleChoi, Heejeong, Il Young Ryu, Inkyu Choi, Sultan Ullah, Hee Jin Jung, Yujin Park, Yeongmu Jeong, YeJi Hwang, Sojeong Hong, In-Soo Yoon, and et al. 2021. "Novel Anti-Melanogenic Compounds, (Z)-5-(Substituted Benzylidene)-4-thioxothiazolidin-2-one Derivatives: In Vitro and In Silico Insights" Molecules 26, no. 16: 4963. https://doi.org/10.3390/molecules26164963