
Lithocholic Acid Induces miR21, Promoting PTEN Inhibition via STAT3 and ERK-1/2 Signaling in Colorectal Cancer Cells

 ,
,  , and
, and
Abstract
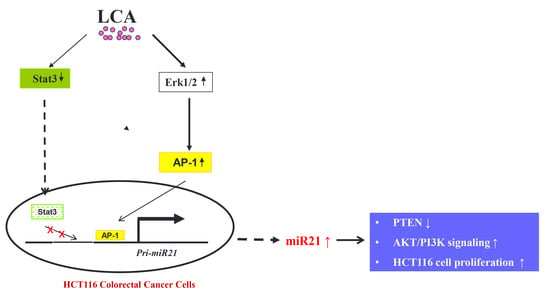
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. LCA Induces miR21 Expression in CRC Cell Lines
2.2. AP-1 Transcription Factor Is Involved in LCA-Induced miR21 Promoter Activity
2.3. Erk1/2 Signal Is Involved in LCA-Induced miR21 Expression through AP-1 Transcription Factor Activation
2.4. Role of STAT3 in LCA-Induced miR21
2.5. LCA Inhibits the PTEN Expression Due to LCA-Induced miR21 in HCT116 Cells
2.6. LCA Enhances the Proliferation of CRC HCT116 Cells via miR21 Upregulation
3. Discussion
4. Materials and Methods
4.1. Cell Culture Conditions and Materials
4.2. miR21 Expression Using Real-Time PCR
4.3. PTEN Transcriptional Expression
4.4. miR21 Promoter Construction
4.5. miR21 Promoter Deletion Mutant Construction
4.6. Promoter Activity Assay
4.7. Western Blot Analysis
4.8. Transfection
4.9. Proliferation Assay
4.10. Densitometric Analysis
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chan, J.; Krichevsky, A.; Kosik, K. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005, 65, 6029–6033. [Google Scholar] [CrossRef] [Green Version]
- Volinia, S.; Calin, G.; Liu, C.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A micro-RNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.-H.; Tsao, C.-J. Emerging role of microRNA-21 in cancer (Review). Biomed. Rep. 2016, 5, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Zhao, L.; Chen, Y.; He, T.; Chen, X.; Mao, J.; Li, C.; Lyu, J.; Meng, Q.H. MicroRNA-21 promotes proliferation, migration, and invasion of colorectal cancer, and tumor growth associated with down-regulation of sec23a expression. BMC Cancer 2016, 16, 605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krichevsky, A.M.; Gabriely, G. miR-21: A small multi-faceted RNA. J. Cell. Mol. Med. 2009, 13, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Cekaite, L.; Eide, P.W.; Lind, G.E.; Skotheim, R.I.; Lothe, R.A. MicroRNAs as growth regulators, their function and biomarker status in colorectal cancer. Oncotarget 2016, 7, 6476–6505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mima, K.; Nishihara, R.; Yang, J.; Dou, R.; Masugi, Y.; Shi, Y.; da Silva, A.; Cao, Y.; Song, M.; Nowak, J. MicroRNA MIR21 (miR-21) and PTGS2 expression in colorectal cancer and patient survival. Clin. Cancer Res. 2016, 22, 3841–3848. [Google Scholar] [CrossRef] [Green Version]
- Ding, T.; Cui, P.; Zhou, Y.; Chen, C.; Zhao, J.; Wang, H.; Guo, M.; He, Z.; Xu, L. Antisense Oligonucleotides against miR-21 Inhibit the Growth and Metastasis of Colorectal Carcinoma via the DUSP8 Pathway. Mol. Ther. Nucleic Acids 2018, 13, 244–255. [Google Scholar] [CrossRef] [Green Version]
- Dixit, A.K.; Sarver, A.E.; Yuan, Z.; George, J.; Barlass, U.; Cheema, H.; Sareen, A.; Banerjee, S.; Dudeja, V.; Dawra, R.; et al. Comprehensive analysis of microRNA signature of mouse pancreatic acini: Overexpression of miR-21-3p in acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G974–G980. [Google Scholar] [CrossRef] [Green Version]
- Krattinger, R.; Boström, A.; Lee, S.M.L.; Thasler, W.E.; Schiöth, H.B.; Kullak-Ublick, G.A.; Mwinyi, J. Chenodeoxycholic acid significantly impacts the expression of miRNAs and genes involved in lipid, bile acid and drug metabolism in human hepatocytes. Life Sci. 2016, 156, 47–56. [Google Scholar] [CrossRef]
- Fujita, S.; Ito, T.; Mizutani, T.; Minoguchi, S.; Yamamichi, N.; Sakurai, K.; Iba, H. miR-21 gene expression triggered by AP-1 is sustained through a double-negative feedback mechanism. J. Mol. Biol. 2008, 378, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Lian, S.; Ung, T.T.; Xia, Y.; Han, J.Y.; Jung, Y.D. Lithocholic Acid Stimulates IL-8 Expression in Human Colorectal Cancer Cells Via Activation of Erk1/2 MAPK and Suppression of STAT3 Activity. J. Cell. Biochem. 2017, 118, 2958–2967. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Sánchez, D.; Arriaga-Canon, C.; Pedroza-Torres, A.; Rosa-Velázquez, I.A.D.L.; González-Barrios, R.; Contreras-Espinosa, L.; Montiel-Manríquez, R.; Castro-Hernández, C.; Fragoso-Ontiveros, V.; Álvarez-Gómez, R.M.; et al. The Promising Role of miR-21 as a Cancer Biomarker and Its Importance in RNA-Based Therapeutics. Mol. Ther. Nucleic Acids 2020, 20, 409–420. [Google Scholar] [CrossRef]
- Wu, K.; Li, L.; Li, S. Circulating microRNA-21 as a biomarker for the detection of various carcinomas: An updated meta-analysis based on 36 studies. Meta-Anal. Tumour Biol. 2015, 36, 1971–1982. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Ung, T.T.; Kim, N.H.; Jung, Y.D. Role of bile acids in colon carcinogenesis. World J. Clin. Cases 2018, 6, 577–588. [Google Scholar] [CrossRef]
- Yuan, T.; Ni, Z.; Han, C.; Min, Y.; Sun, N.; Liu, C.; Shi, M.; Lu, W.; Wang, N.; Du, F.; et al. SOX2 interferes with the function of CDX2 in bile acid-induced gastric intestinal metaplasia. Cancer Cell Int. 2019, 19, 24. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.M.; Afonso, M.B.; Simão, A.L.; Borralho, P.M.; Rodrigues, C.M.P.; Castro, R.E. Inhibition of NF-κB by deoxycholic acid induces miR-21/PDCD4- dependent hepatocellular apoptosis. Sci. Rep. 2015, 5, 17528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calo, N.; Ramadori, P.; Sobolewski, C.; Romero, Y.; Maeder, C.; Fournier, M.; Rantakari, P.; Zhang, F.-P.; Poutanen, M.; Dufour, J.-F.; et al. Stress-activated miR-21/miR-21* in hepatocytes promotes lipid and glucose metabolic disorders associated with high-fat diet consumption. Gut 2016, 65, 1871–1881. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Hwang, S.H.; Cho, H.H.; Shin, K.K.; Bae, Y.C.; Jung, J.S. MicroRNA 21 regulates the proliferation of human adipose tissue-derived mesenchymal stem cells and high-fat diet-induced obesity alters microRNA 21 expression in white adipose tissues. J. Cell. Physiol. 2012, 227, 183–193. [Google Scholar] [CrossRef]
- Wu, H.; Ng, R.; Chen, X.; Steer, C.J.; Song, G. MicroRNA-21 is a potential link between non-alcoholic fatty liver disease and hepatocellular carcinoma via modulation of the HBP1-p53-Srebp1c pathway. Gut 2016, 65, 1850–1860. [Google Scholar] [CrossRef] [Green Version]
- Nakata, K.; Sugi, Y.; Narabayashi, H.; Kobayakawa, T.; Nakanishi, Y.; Tsuda, M.; Hosono, A.; Kaminogawa, S.; Hanazawa, S.; Takahashi, K. Commensal microbiota-induced microRNA modulates intestinal epithelial permeability through the small GTPase ARF4. J. Biol. Chem. 2017, 292, 15426–15433. [Google Scholar] [CrossRef] [Green Version]
- Ajouz, H.; Mukherji, D.; Shamseddine, A. Secondary bile acids: An underrecognized cause of colon cancer. World J. Surg. Oncol. 2014, 12, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funabashi, M.; Grove, T.L.; Wang, M.; Varma, Y.; McFadden, M.E.; Brown, L.C.; Guo, C.; Higginbottom, S.; Almo, S.C.; Fischbach, M.A. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature 2020, 582, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, W.D.; Wang, Y.D. The roles of the gut microbiota–miRNA interaction in the host pathophysiology. Mol. Med. 2020, 26, 101. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.K.; Park, J.S.; Park, J.H.; Kim, M.H.; Kim, H.D.; Bae, W.K.; Chung, I.J.; Shin, B.A.; Jung, Y.D. Lithocholic acid upregulates uPAR and cell invasiveness via MAPK and AP-1 signaling in colon cancer cells. Cancer Lett. 2010, 290, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, J.; Wang, H.; Shi, J.; Wu, K.; Liu, S.; Liu, Y.; Wu, J. HCV-Induced miR-21 Contributes to Evasion of Host Immune System by Targeting MyD88 and IRAK1. PLoS Pathog. 2013, 9, e1003248. [Google Scholar] [CrossRef] [PubMed]
- Mercado-Pimentel, M.E.; Onyeagucha, B.C.; Li, Q.; Pimentel, A.C.; Jandova, J.; Nelson, M.A. The S100P/RAGE signaling pathway regulates expression of microRNA-21 in colon cancer cells. FEBS Lett. 2015, 589, 2388–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, Y.; Radde, B.N.; Litchfield, L.M.; Ivanova, M.M.; Prough, R.A.; Clark, B.J.; Doll, M.A.; Hein, D.W.; Klinge, C.M. Dehydroepiandrosterone Activation of G-protein-coupled Estrogen Receptor Rapidly Stimulates MicroRNA-21 Transcription in Human Hepatocellular Carcinoma Cells. J. Biol. Chem. 2015, 290, 15799–15811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, T.J.; John, S. Signal transducer and activator of transcription (STAT) signalling and T-cell lymphomas. Immunology 2005, 114, 301–312. [Google Scholar] [CrossRef]
- Zhang, H.-F.; Lai, R. STAT3 in Cancer—Friend or Foe? Cancers 2014, 6, 1408–1440. [Google Scholar] [CrossRef] [Green Version]
- Löffler, D.; Brocke-Heidrich, K.; Pfeifer, G.; Stocsits, C.; Hackermüller, J.; Kretzschmar, A.K.; Burger, R.; Gramatzki, M.; Blumert, C.; Bauer, K.; et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 2007, 110, 1330–1333. [Google Scholar] [CrossRef] [Green Version]
- Tscherner, A.; Brown, A.C.; Stalker, L.; Kao, J.; Dufort, I.; Sirard, M.-A.; LaMarre, J. STAT3 signaling stimulates miR-21 expression in bovine cumulus cells during in vitro oocyte maturation. Sci. Rep. 2018, 8, 11527. [Google Scholar] [CrossRef] [Green Version]
- Fits, L.v.d.; Kester, M.S.v.; Qin, Y.; Out-Luiting, J.J.; Smit, F.; Zoutman, W.H.; Willemze, R.; Tensen, C.P.; Vermeer, M.H. MicroRNA-21 Expression in CD4 þ T Cells Is Regulated by STAT3 and Is Pathologically Involved in Se´zary Syndrome. J. Investig. Dermatol. 2011, 131, 762–768. [Google Scholar] [CrossRef] [Green Version]
- Gutsaeva, D.R.; Thounaojam, M.; Rajpurohit, S.; Powell, F.L.; Martin, P.M.; Goei, S.; Duncan, M.; Bartoli, M. STAT3-mediated activation of miR-21 is involved in downregulation of TIMP3 and neovascularization in the ischemic retina. Oncotarget 2017, 8, 103568–103580. [Google Scholar] [CrossRef]
- Ou, H.; Li, Y.; Kang, M. Activation of miR-21 by STAT3 Induces Proliferation and Suppresses Apoptosis in Nasopharyngeal Carcinoma by Targeting PTEN Gene. PLoS ONE 2014, 9, e109929. [Google Scholar] [CrossRef]
- Ohno, M.; Natsume, A.; Kondo, Y.; Iwamizu, H.; Motomura, K.; Toda, H.; Ito, M.; Kato, T.; Wakabayashi, T. The Modulation of MicroRNAs by Type I IFN through the Activation of Signal Transducers and Activators of Transcription 3 in Human Glioma. Mol. Cancer Res. 2009, 7, 2022–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.W.; Zeng, H.S. Regulation of JAK/STAT signal pathway by miR-21 in the pathogenesis of juvenile idiopathic arthritis. World J. Pediatrics 2020, 16, 502–513. [Google Scholar] [CrossRef] [Green Version]
- De Jong, P.R.; Mo, J.H.; Harris, A.R.; Lee, J.; Raz, E. STAT3: An Anti-Invasive Factor in Colorectal Cancer. Cancers 2014, 6, 1394–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musteanu, M.; Blaas, L.; Mair, M.; Schlederer, M.; Bilban, M.; Tauber, S.; Esterbauer, H.; Mueller, M.; Casanova, E.; Kenner, L.; et al. Stat3 is a negative regulator of intestinal tumor progression in Apc(Min) mice. Gastroenterology 2010, 138, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Long, Z.; Wu, J.; Cai-Hong, Y.N.W.; Zhou, Y. MiR-374b Promotes Proliferation and Inhibits Apoptosis of Human GIST Cells by Inhibiting PTEN through Activation of the PI3K/Akt Pathway. Mol. Cells 2018, 41, 532–544. [Google Scholar] [PubMed]
- Wu, Y.-R.; Qi, H.-J.; Deng, D.-F.; Luo, Y.-Y.; Yang, S.-L. MicroRNA-21 promotes cell proliferation, migration, and resistance to apoptosis through PTEN/PI3K/AKT signaling pathway in esophageal cancer. Tumor Biol. 2016, 37, 12061–12070. [Google Scholar] [CrossRef]
- Liu, H.-Y.; Zhang, Y.-Y.; Zhu, B.-L.; Feng, F.-Z.; Yan, H.; Zhang, H.-Y.; Zhou, B. miR-21 regulates the proliferation and apoptosis of ovarian cancer cells through PTEN/PI3K/AKT. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 4149–4155. [Google Scholar]
- Zheng, Y.; Zhang, D. Micro RNA-21 Targeting PI3K/PTEN/Akt signaling axis regulates multiple biological functions in human lung cancer. Eur. Respir. J. 2018, 52, PA2847. [Google Scholar]
- Hao, X.-J.; Xu, C.-Z.; Wang, J.-T.; Li, X.-J.; Wang, M.-M. miR-21 promotes proliferation and inhibits apoptosis of hepatic stellate cells through targeting PTEN/PI3K/AKT pathway. J. Recept. Signal Transduct. 2018, 38, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Anwer, M.S. Intracellular signaling by bile acids. J. Biosci. 2012, 20, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soliman, M.; Kim, D.-S.; Park, J.-G.; Kim, J.-Y.; Alfajaro, M.M.; Baek, Y.-B.; Cho, E.-H.; Park, C.-H.; Kang, M.-I.; Park, S.-I.; et al. Phosphatidylinositol 3-Kinase/Akt and MEK/ERK Signaling Pathways Facilitate Sapovirus Trafficking and Late Endosomal Acidification for Viral Uncoating in LLC-PK Cells. J. Virol. 2018, 92, e01674-18. [Google Scholar] [CrossRef] [Green Version]
- Raufman, J.-P.; Shant, J.; Guo, C.Y.; Roy, S.; Cheng, K. Deoxycholyltaurine rescues human colon cancer cells from apoptosis by activating EGFR-dependent PI3K/Akt signaling. J. Cell. Physiol. 2008, 215, 538–549. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.; Ung, T.T.; Li, S.; Lian, S.; Xia, Y.; Park, S.Y.; Jung, Y.D. Metformin inhibits lithocholic acid-induced interleukin 8 upregulation in colorectal cancer cells by suppressing ROS production and NF-kB activity. Sci. Rep. Vol. 2019, 9, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.-T.; Ung, T.-T.; Li, S.; Sah, D.K.; Park, S.-Y.; Lian, S.; Jung, Y.-D. Lithocholic Acid Induces miR21, Promoting PTEN Inhibition via STAT3 and ERK-1/2 Signaling in Colorectal Cancer Cells. Int. J. Mol. Sci. 2021, 22, 10209. https://doi.org/10.3390/ijms221910209
Nguyen T-T, Ung T-T, Li S, Sah DK, Park S-Y, Lian S, Jung Y-D. Lithocholic Acid Induces miR21, Promoting PTEN Inhibition via STAT3 and ERK-1/2 Signaling in Colorectal Cancer Cells. International Journal of Molecular Sciences. 2021; 22(19):10209. https://doi.org/10.3390/ijms221910209
Chicago/Turabian StyleNguyen, Thinh-Thi, Thuan-Trong Ung, Shinan Li, Dhiraj Kumar Sah, Sun-Young Park, Sen Lian, and Young-Do Jung. 2021. "Lithocholic Acid Induces miR21, Promoting PTEN Inhibition via STAT3 and ERK-1/2 Signaling in Colorectal Cancer Cells" International Journal of Molecular Sciences 22, no. 19: 10209. https://doi.org/10.3390/ijms221910209